Caratterizzazione di film di Carbonio
Amorfo
2000-2001
Il Carbonio
Amorfo è un materiale che sta destando vivo interesse nella Fisica degli Stati
Condensati, sia nel campo della ricerca fondamentale, per le sue peculiari
caratteristiche strutturali ed opto-elettroniche, sia per le sue promettenti
applicazioni tecnologiche, che spaziano dall’elettronica alla meccanica ed alla
biomeccanica.
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
Il
fenomeno fisico |
|
|
|
||
|
|
||
Storicamente,
nella Fisica degli stati condensati, il concetto di cristallo è sempre stato
considerato come sinonimo di materiale nello stato solido. Ogni corso di fisica
della Materia inizia la sua trattazione dalla definizione di reticolo
periodico, di simmetria, di invarianza e di tutte le tecniche matematiche
(teoria dei gruppi, trasformate di Fourier, etc) per
mezzo delle quali si sono potute indagare le proprietà basilari dei cristalli.
I materiali disordinati, come ad esempio quelli vetrosi o amorfi, sono stati
per molto tempo considerati "indegni" della qualifica di materia nello
stato solido, ma piuttosto come nello stato liquido in una condizione
particolare di alta densità.
A rafforzare
quest’ottica è il fatto che un materiale disordinato si trova in una
configurazione metastabile rispetto ad una possibile fase cristallina, che
rappresenta lo stato termodinamico di equilibrio stabile, al livello energetico
fondamentale; le sue particelle costituenti non sono disposte in uno stato di
minimo energetico e quindi la sua configurazione strutturale è mutevole come
quella di un liquido, seppur in archi di tempo enormemente più lunghi di quelli
dei fluidi propriamente detti.
A partire
dalla prima metà degli anni ’80 però i materiali amorfi hanno acquisito
"sul campo" (cioè in numerose applicazioni tecnologiche: dalle celle
solari alla xerografia, fino ad applicazioni nel campo dell’elettronica) una
"rispettabilità" nell’ambito dei materiali allo stato solido. Già dal
punto di vista tecnologico si è realizzato che la periodicità non è un
prerequisito fondamentale per le caratteristiche di durezza e resistenza
meccaniche tipiche dei solidi. Infine la stessa argomentazione sulla metastabilità degli aggregati disordinati della materia,
benché inoppugnabile, è stata da alcuni studiosi messa in discussione come una
argomentazione più "accademica" che sostanziale: la fase cristallina
è in molti casi cineticamente inaccessibile al solido
disordinato, e quindi questo può persistere indefinitamente nella fase amorfa.
Per esempio la configurazione a più bassa energia degli atomi di carbonio non è
il diamante, ma la grafite, quindi lo stesso diamante (considerato
"il" solido per eccellenza per le sue eccezionali caratteristiche
meccaniche) si trova in una configurazione metastabile e in linea teorica può
transire ad un stato ad energia più bassa. In pratica però, in condizioni di
pressione e temperatura standard (STP), un diamante persiste indefinitamente
nel suo stato. Lo stesso ragionamento vale per molti solidi disordinati. In
base a queste considerazioni è lecito affermare che la periodicità strutturale
non è sinonimo di "solidità".
La condizione
di disordine strutturale in un solido non deve fare pensare ad una situazione
in cui gli atomi costituenti il materiale siano disposti in modo completamente
casuale. Questo modello si adatta meglio alla descrizione di un gas o di un
fluido. Un solido viene definito disordinato in quanto nella sua struttura
reticolare è assente l’ordine a lungo range,
ossia la periodicità e la simmetria. Ciò nonostante l’ordine a corto (e
talvolta medio) range viene preservato, perché
anche in un amorfo i siti reticolari sono correlati localmente da legami
chimici della stessa natura di quelli del corrispondente stato cristallino,
salvo piccole distorsioni nella distanza o nell’angolo diedro tra atomi primi
vicini.
Questo tipo di
struttura deve essere descritto con modelli diversi da quello classico del
reticolo periodico; quelli che vanno sotto il nome di Random Close Packing, Random Coil e Continuous
Random Network modellizzano una struttura globalmente disordinata
rispettivamente mediante l’impacchettamento casuale di sfere rigide,
l’intrecciarsi disordinato di catene e strutture lineari, e un reticolo
periodico nel quale le distanze e gli angoli tra primi vicini abbiano
distribuzioni statistiche.
Nello studio
della dinamica degli elettroni all’interno di materiali di tipo cristallino
sono di fondamentale importanza due tipi di transizione: la transizione di
Bloch e di Mott.
La transizione
di Bloch è la più rilevante transizione isolante/conduttore in un solido
cristallino; in base a questa teoria, dalla definizione di una legge di
dispersione che lega l’energia (E) al momento cristallino k
dell’elettrone:
E = En (k)
si giunge alla
definizione di bande energetiche nel materiale; ogni banda può contenere un
numero massimo di stati elettronici estesi, cioè delocalizzati, per effetto del
principio di esclusione di Pauli. La formazione di stati estesi è
energeticamente favorita da un guadagno energetico di delocalizzazione. Da
considerazioni sulla struttura delle bande e sul numero di elettroni in esse
presenti consegue la distinzione tra cristalli conduttori ed isolanti.
La transizione
di Mott implica una rottura della struttura in
bande succitata, inserendo l’effetto di un’interazione a molti corpi (la
repulsione elettrostatica degli elettroni) che permette di distinguere
energeticamente diverse configurazioni elettroniche equivalenti tra loro in un
approccio a particelle indipendenti (one-electron theory). Nell’esempio della figura seguente, la
configurazione elettronica in figura 1.a è energeticamente favorita rispetto a
quella riportata in 1.b.
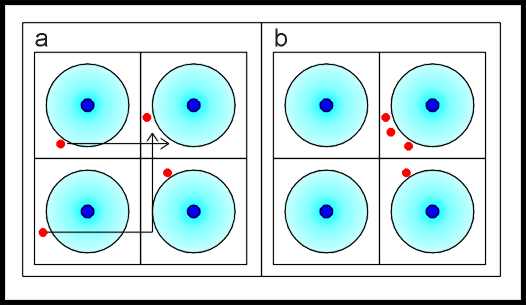
|
Transizione di Mott |
fig.1 |
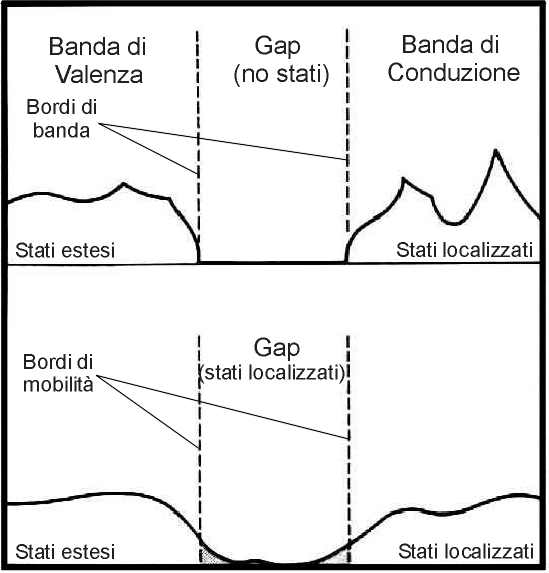
In un solido
amorfo la più rilevante transizione isolante/conduttore è la transizione di
Anderson. Si basa sulla distinzione tra stati delocalizzati e stati
localizzati per effetto del disordine strutturale. Al caso di materiale
isolante corrisponde la localizzazione degli stati elettronici, mentre alla
presenza di stati estesi corrisponde una situazione di materiale conduttivo.
Si noti che mentre nei cristalli gli stati localizzati sono
estrinseci, cioè introdotti da impurezze
chimiche ed imperfezioni strutturali, negli amorfi gli stati localizzati sono intrinseci
alla struttura del materiale, essendo determinati dal disordine strutturale, e
formano un spettro continuo di stati.
Se la
configurazione delle buche di potenziale in un cristallo può essere
schematizzata in figura 2.a, in un materiale disordinato la situazione si
modifica nel modo riportato in figura 2.b.
|
Transizione di Anderson |
fig. 3 |
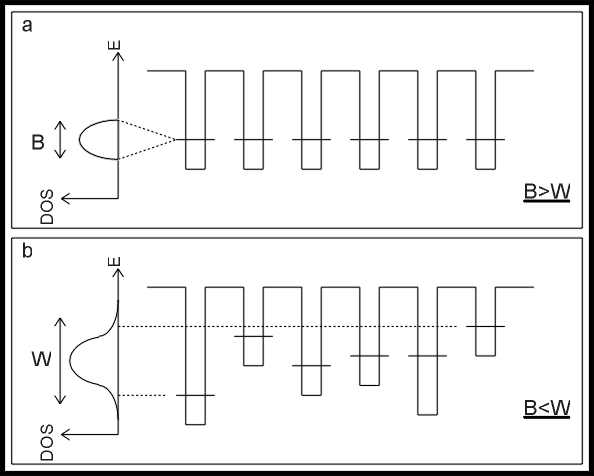
Come mostrato
in figura 3.b, la profondità delle buche di potenziale ha una certa
distribuzione la cui larghezza caratteristica è correlata al parametro di
disordine (W). Questo parametro deve essere confrontato con il guadagno
energetico di delocalizzazione (B): cruciale è il rapporto tra le due
grandezze caratteristiche.
Quando esso è
sufficientemente grande, tutti gli stati nella banda di valenza sono
localizzati a causa del disordine strutturale. Anche quando non è
sufficientemente grande da rendere localizzati tutti gli stati nella banda di
valenza, una parte degli elettroni nel materiale si localizza comunque: si
tratta, nel caso dei semiconduttori, di stati sulle code di banda, che
penetrano nella gap proibita "smussando" e rendendo indefiniti i
bordi degli stati a più alta energia della banda di valenza e di quelli a più
bassa energia della banda di conduzione, come mostrato in figura 4.
|
Stati elettronici localizzati |
fig. 4 |
Tutti i
semiconduttori, sia cristallini che amorfi, esibiscono uno spigolo (o spalla)
di assorbimento. In corrispondenza di esso il coefficiente di assorbimento
ottico incrementa di molti ordini di grandezza su un relativamente ristretto range di energie fotoniche. Questo netto incremento
definisce la "gap ottica" per le transizioni ottiche tra gli stati
occupati della banda di valenza e quelli non occupati della banda di
conduzione. Questo è un parametro critico per molte applicazioni ottiche del
semiconduttore ed un test sperimentale per i calcoli teorici sulla struttura a
bande del semiconduttore stesso. La dipendenza energetica del coefficiente di
assorbimento ottico in prossimità della gap ottica è anch’essa di grande
interesse, dal momento che fornisce molte informazioni sul meccanismo di
assorbimento. Esistono tre picchi di assorbimento per un semiconduttore, che
corrispondono alla fisica della transizione ottica: lo spigolo diretto
di un semiconduttore cristallino a gap diretta, lo spigolo indiretto di
un semiconduttore cristallino a gap indiretta e lo spigolo non-diretto
di un semiconduttore amorfo. La peculiarità di quest’ultimo tipo di
assorbimento è dovuta al fatto che l’assenza di invarianza traslazionale nella
struttura disordinata fa sì che il momento cristallino (k) non
sia più un buon numero quantico; le transizioni ottiche possono dunque avvenire
con la conservazione dell’energia come unico vincolo. Il modello che descrive
questo tipo di transizioni va sotto il nome di modello a Bande Indipendenti.
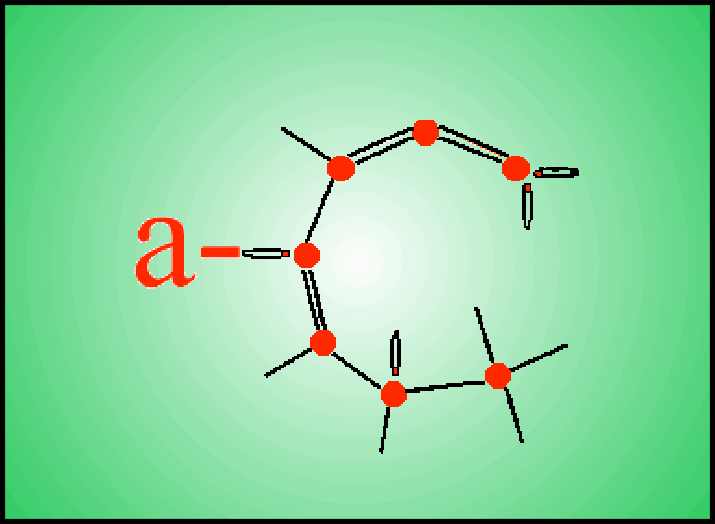
Il Carbonio ha
numero atomico 6 e la sua struttura elettronica è:
[He]2s22p2
Gli elettroni
di valenza dell’elemento sono quattro e possono dar luogo a diversi tipi di
legame chimico dal momento che uno degli elettroni nello stato s ha la
possibilità, nella formazione del legame, di passare allo stato p e che
gli orbitali s e p hanno la possibilità di ibridizzarsi tra loro
in modi differenti: si possono formare legami tipo sp1, sp2
ed sp3, nel caso in cui un orbitale s si ibridizzi
rispettivamente con 1, 2 o 3 orbitali p.
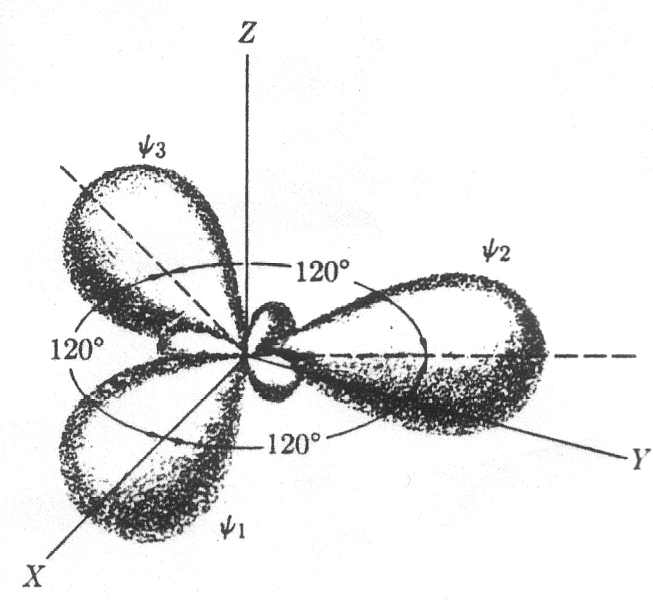
Gli orbitali sp2
risultano dall’ibridazione di un orbitale s con due orbitali p
(convenzionalmente px e py); formano tre legami tipo s
, cioè diretti lungo il segmento congiungente i nuclei dei due atomi
legati, disposti complanarmente ad angoli di 120°; il
quarto orbitale è il restante pz, che si sviluppa
perpendicolarmente al piano succitato, formando un legame tipo p , non diretto lungo la congiungente i
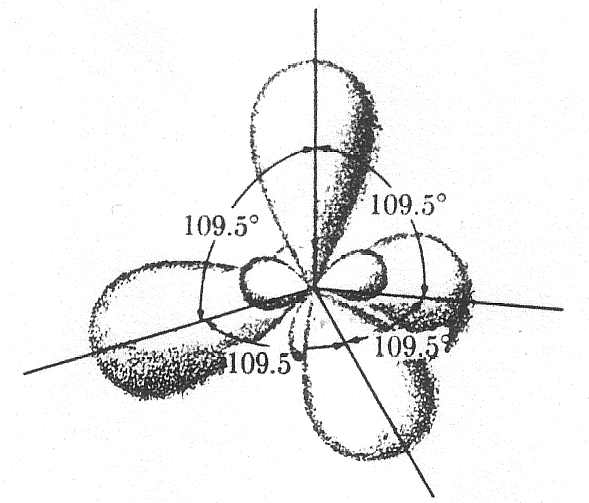
due atomi e meno saldo energeticamente. Gli orbitali sp3
risultano dall’ibridazione di un orbitale s con tre orbitali p (px, py
e pz) formano quattro legami tipo s
disposti ad angoli di 109.5°. La struttura di questi stati è illustrata
in figura 5.
|
|
|
|
Ibridazione sp2 |
Ibridazione sp3 |
|
fig. 5 |
|
Il Carbonio si
presenta in tre forme allotropiche: diamante, grafite ed amorfo.
In ciascuna di queste tre diverse configurazioni sono coinvolti diversi tipi di
legame che ne determinano le proprietà sia meccaniche che elettroniche.
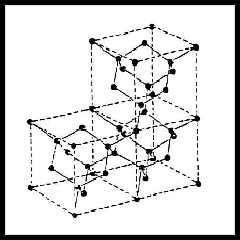
- Il diamante è un cristallo covalente a coordinazione tetraedrica;
la sua struttura cristallina è cubica a facce centrate in cui ad ogni
punto reticolare è associata una base di due atomi di Carbonio nello
coordinate (0,0,0) e (¼,¼,¼); la costante
reticolare è 3.57 Å e la distanza dai primi vicini è 1.54 Å; ogni atomo di
Carbonio è legato alla struttura cristallina mediante quattro orbitali
ibridi sp3. Il diamante è in assoluto il più duro
materiale presente in natura grazie al carattere covalente del legame e
dal punto di vista elettronico è un semiconduttore con una gap di 5.45 eV.
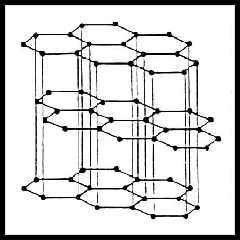
- La
grafite è un cristallo formato da un
insieme di piani reticolari (detti anche basali) nei quali gli
atomi formano una rete esagonale connessa da orbitali ibridi sp2
rafforzati dal legame risonante dei restanti orbitali p, che sono
quindi delocalizzati su tutta la struttura planare; la grande
mobilità degli elettroni in questi stati conferisce alla sostanza una
conduzione di carattere metallico. I piani basali costituiscono dei
complessi chimicamente saturi, e rappresentano la configurazione
energeticamente più stabile per il Carbonio. Piani diversi sono tenuti
insieme tra loro da forze di Van der Waals, molto più deboli di quelle di valenza. È questo
a conferire una scarsissima durezza al materiale, che è uno dei più
morbidi solidi cristallini conosciuti.
- Il
Carbonio amorfo è la versione strutturalmente
disordinata in cui può solidificare il materiale. In questa struttura
coesistono in diverse percentuali i tre diversi tipi di legame chimico cui
può dar luogo il Carbonio: sp1, sp2 ed sp3.
Il materiale si presenta quindi come una sorta di "ibrido" tra
le due configurazioni succitate, che hanno proprietà sia elettroniche che
meccaniche addirittura antitetiche. Appare dunque evidente come questo
materiale possa presentarsi in numerose varianti strutturali, che coprono
una vasta gamma di proprietà fisiche.
|
|
|
|
|
Diamante |
Grafite |
Carbonio Amorfo |
|
fig. 6 |
||

Una vasta
gamma di tecniche di deposizione porta alla produzione di Carbonio amorfo in
molte varianti strutturali, che si differenziano radicalmente tra loro sia
nelle proprietà meccaniche che in quelle opto-elettroniche. Il rapporto tra
siti coordinati mediante legami tetraedrici sp3 (diamondlike) e siti di tipo trigonale planare sp2
(grafitici) ed il contenuto di Idrogeno (o di altri elementi droganti, come
l’Azoto, il Fluoro, etc.) sono i due parametri basilari per interpretare le
proprietà dei film sottili di Carbonio amorfo. È utile pertanto riassumere
schematicamente la gamma di varietà di questo materiale nel semplice diagramma
riportato in figura 7. Il Carbonio amorfo fortemente grafitizzato si localizza
nell’angolo in basso a sinistra; i polimeri sulla destra; il Carbonio amorfo
tetraedrico in prossimità del vertice in alto. Nelle zone centrali si situano
il Carbonio amorfo idrogenato, tetraedrico (ta-C:H) o meno (ta-C).
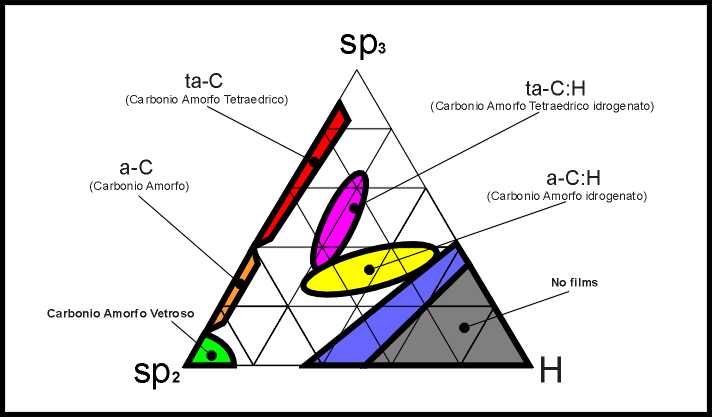
|
Catalogazione schematica delle varietà di Carbonio Amorfo |
fig. 7 |
Le
caratteristiche meccaniche ed elettroniche del materiale dipendono pesantemente
dalle condizioni in cui esso viene deposto, dal momento che queste determinano
direttamente i parametri succitati. Le principali tecniche di deposizione del
materiale sono schematizzate in figura 8.
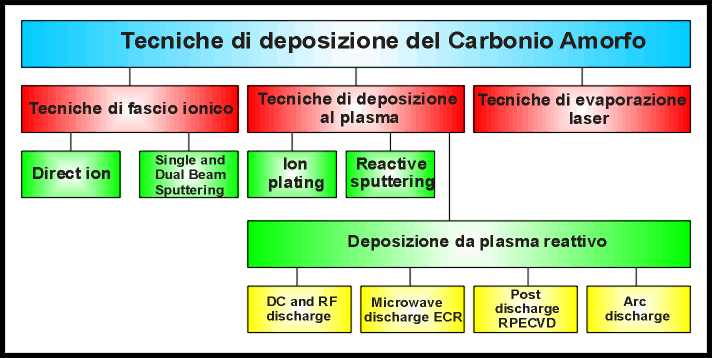
|
Tecniche di deposizione |
fig. 8 |
Misure PAS
e PDS su carbonio Amorfo
Mediante le
tecniche fotoacustiche PAS e PDS si sono studiate le regioni
spettrali nel visibile di bassi assorbimenti ottici di film sottili di Carbonio
Amorfo deposti mediante sputtering reattivo presso il
Dipartimento di Fisica del Politecnico di Torino e mediante tecnica FCVA presso
il Dipartimento di Ingegneria dell’Università di Cambridge. Lo studio dei bassi
assorbimenti ottici ha lo scopo di investigare la densità di stati elettronici p e p* in
prossimità del livello di Fermi.
Nelle figure
seguenti sono riportati i risultati delle misure PAS e PDS effettuate congiuntamente.
|
|
|
|
Campione n.1 ( a-C:H:N ) |
Campione n.2 ( ta-C ) |
|
|
|
|
Campione n.3 ( ta-C ) |
Campione n.4 ( ta-C:N ) |
|
figg. 9, 10, 11, 12 |
|
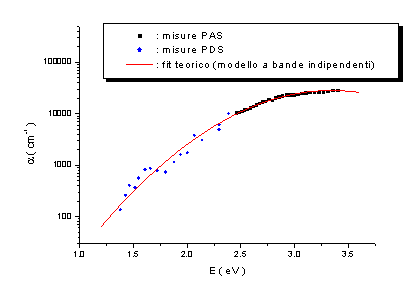
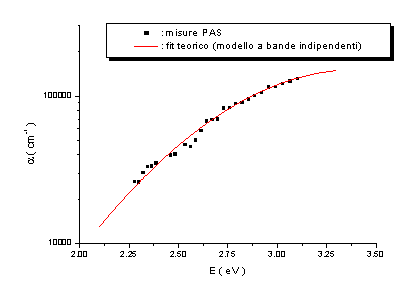
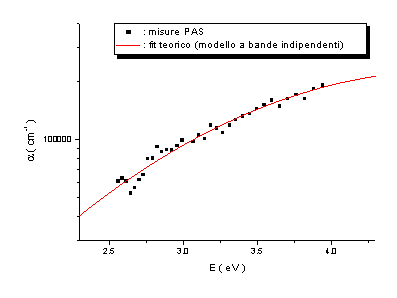
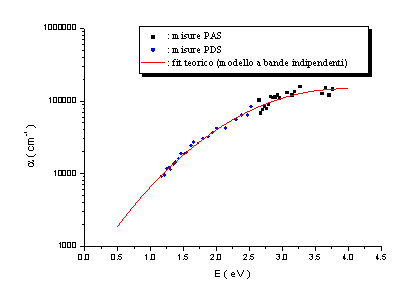
Le misure PAS
e PDS effettuate presentano un accordo soddisfacente ed evidenziano altresì come
la tecnica di Deflessione Fototermica sia più sensibile rispetto alla
Fotoacustica. Dall’analisi di questi spettri è stato possibile formulare alcune
ipotesi sulla densità di stati elettronici p e
p* in prossimità del
livello di Fermi, ipotizzando che i bassi assorbimenti siano dovuti a transizioni p ® p*, come schematizzato in figura 13.
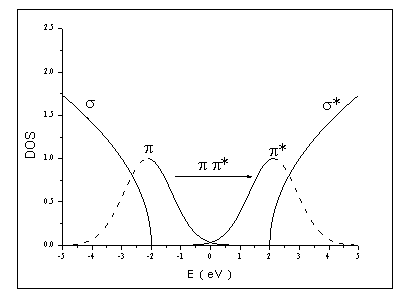
|
Densità di stati p e p* |
fig. 13 |
Dall’analisi
degli spettri di assorbimento è emerso che tali densità di stati possono essere
modellizzate con gaussiane simmetriche rispetto al livello di Fermi. Ponendo
per comodità EF=0 eV, queste sono
centrate in ±Ep ed hanno larghezza comune
s.
Si sono ottenuti valori di Ep
compresi tra 1.7 e 2.6 eV, e valori di s
compresi tra 0.4 e 0.9 eV.
Partecipanti alla ricerca: C. Manfredotti, E. Vittone, F. Fizzotti, P. Olivero